Monday's journal club was on this Wang et al. 1997 paper. They took the hERG gene, which was thought to be responsible for the rapid delayed rectifier potassium current in humans, and put it into unfertilised frog eggs, then used patch clamping to find out its properties.
I think a lot about how the membrane potential is affected by the currents that are flowing across it, but sometimes it is good to remember that the currents themselves are modified by the membrane voltage. In cardiac models, the currents tend to be modelled as:
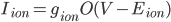
where 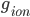 is the conductance,
is the conductance, 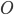 is the proportion of channels that are active,
is the proportion of channels that are active, 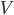 is the membrane voltage, and
is the membrane voltage, and 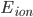 the reversal potential. is often affected by the membrane potential, as well, so the membrane voltage plays a key role in regulating current flow.
the reversal potential. is often affected by the membrane potential, as well, so the membrane voltage plays a key role in regulating current flow.
Usually, the voltage across the membrane of a cardiac cell looks a bit like this:

If we're doing a patch clamp then we stick an electrode to the outside of the cell, which means that we can control what the membrane potential does. We could make it do this:

... or use the same shape, but don't bring the voltage up as far:

In the case of hERG, this lets us see what happens when the current is activated, because hERG switches on at high voltages. By comparing the activation time at lots of different potential steps, you can begin to understand the activation kinetics of the channel. In this paper, they were able to put together a model of what happens, by taking models with various numbers of closed states and comparing their predictions to the real data.
Activation kinetics are important, but so are deactivation kinetics. The hERG channels close when the membrane potential decreases, so if we activate them and then let the voltage drop down again, we should be able to see what happens when they close:

Again, we can try lots of different voltages:

... and from this, build up a model of the deactivation of this current. Inactivation (which is distinct from deactivation, because it stops the channels from being openable for a period of time) happens very quickly, so they had to cut open the cells for their patch clamp. I'd never come across this method before, but I came across quite a good review article.
In addition to creating a mathematical model of this current, the authors also talk a little bit about possible molecular reasons for the behaviour of the channel. This isn't something I've seen a lot in electrophysiology modelling papers, and I think it's really important to keep an eye on the fact that in addition to being a model component, what we're modelling is a real, physical protein, and mechanistic understanding of how that works can feed back into modelling work.








